Cardiomyopathy.
PKGhatak, MD
Cardiomyopathy is a weakened condition of the Heart Muscles and the heart chambers increase in size and subsequently the heart fails. It is suspected that 1 in 5,00 adult people may have cardiomyopathy without being aware of it. People seek medical attention only when cardiomyopathy becomes symptomatic. The initial symptoms are shortness of breath and a marked decrease in energy level to carry on the usual work.
There are wide varieties of conditions that lead to cardiomyopathy and based on pathophysiology the disease is classified as Dilated, Hypertrophic, Restricted, and Arrhythmogenic Cardiomyopathy.
Cardiomyopathy also happens due to inherited defects of genes, producing structural changes in the heart muscle or metabolic defects, which cause accumulation of iron and other material in the heart muscles.
I. Dilated Cardiomyopathy:
The heart is enlarged and the muscles are stretched and become thin. Thinned out heart muscles fall behind in pumping blood out of ventricles into the circulation. Accumulated blood causes congestion of the lungs, and produces shortness of breath and in other organs. Dilated cardiomyopathy is diagnosed by clinical examination and confirmed by a chest x-ray and echocardiogram.

A few common causes of dilated myopathy.
1. Virus infection.
Viral myocarditis of a mild nature happens frequently. In most cases, myocarditis resolves spontaneously without any health consequences. The common viruses causing these conditions are Parvovirus B-19, Coxsackie A & B, HIV, Covid-19, Cytomegalovirus, Epstein-Barr virus and Adenoviruses.
Damage to heart muscles in viral infection happens in two stages. In the initial phase, the inflammatory cells infiltrate the heart muscle. Immunocytes release acute phase inflammatory cytokines namely IL-1 beta, TNF alpha, and Interferon alpha and these agents should be sufficient to avert a full blown inflammation, but in some cases, they fail. In the second phase, about 7 to 14 days after the initial symptoms, T-cells accumulate. Vasoconstriction and release of Perforin molecule create multiple pores like apertures in heart muscles. Immunocytes release excess amounts of IL-1, IL-6, CCL5, and TNF beta. Interferon decreases B-Cell activities and facilitates T-Cell activation. Cytokines continued to appear in the heart in response to the presence of viral particles in the myocardium. Because of the close resemblance of the viral antigen to muscle protein, the cytokines make mistakes one for the other. and damages the heart muscle.
A diagnosis of viral cardiomyopathy requires endocardial biopsy and identifying viral antigens. Heart failure and other organ insufficiencies are treated with medications. A cardiac transplant is a definitive treatment.
2. Chagas Disease:
Chagas disease is due to a protozoan parasite, Trypanosoma cruzi. It is an important cause of dilated cardiomyopathy in the Americas. In Central and South American countries, the Chagas disease is endemic. It is estimated that 5 million people are suffering from Chagas disease with an annual rate of infection of 200,000 and annual deaths of 10,000.
About 40,000 people were identified with Chagas disease in the USA among the South American migrant communities in 2019.
The parasite is carried by a bug, Triatoma infestans, commonly known as the kissing bug. The bug bites humans during sleep and leaves a fecal deposit at the site. The contaminated puncture wound, usually on the face or forehead, is the entry point of the parasite. Local inflammation and adenopathy may or may not produce symptoms. About 5 % of victims develop myocarditis. In the chronic phase, 40% develop dilated cardiomyopathy from the continued presence of parasites at the heart and resultant immune inflammation, tissue necrosis and scars replacing heart muscles. Cardiac conduction abnormalities are common and later heart failure predominates.
The diagram shows the life cycle of T. cruzi.
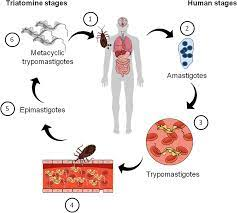
3. Alcoholic Myopathy.
People are well aware of liver disease due to alcohol; however, many will be surprised to learn about 25 % of all cardiomyopathy. Alcohol acts as an important factor in causing cardiomyopathy. About 2% of heavy alcohol users risk cardiac complications. Colored people have more risks of cardiomyopathy than light colored people. Families with Alcoholdehydrogenase deficiency are more likely to have cardiomyopathy. Alcohol is a Mitochondrial poison. As mitochondrial functions falter, acetaldehyde accumulates in the cells and oxygen radicals form as a consequence and the combined effects of these produce cell deaths. Cardiac muscles are replaced by weak scar tissue, and cardiomyopathy develops in the same way as described earlier. There are no distinctive histological characteristics of alcoholic myopathy, however enlarged and disorganized sarcoplasmic reticulum, fat and glycogen deposition and dilated intercalating discs are seen under electron microscopy in addition to various stages of diseased and disappearing mitochondria.
4. Chronic coronary arterial disease and Hypertension.
These two diseases are silent killers of heart muscles. Acute coronary events produce damage to heart muscles, even when emergency Angioplasty is successfully performed and helps to salvage most of the ventricular muscles, some loss is unavoidable. Chronic insufficiency produces almost identical damage but the pace is slow and takes place over a much longer time. Scar formation eventually leads to dilated myopathy and heart failure.
Hypertension produces ventricular muscle hypertrophy initially. If left untreated, the hypertrophic muscles eventually flatten out. A flabby enlarged heart fails. Hypertension is usually associated with coronary artery disease and induces cardiomyopathy but in the absence of coronary artery disease, hypertension alone is an independent cause of cardiomyopathy.
5. Diabetes mellitus.
Diabetes mellitus produces microvascular changes. The exact statistic is variable based on the nature of the inquiry but consistently higher than the controlled groups.
6. Thyroid disorder.
Both hypothyroidism and hyperthyroidism produce Cardiomyopathy but the mechanism is different. Hypofunction of the thyroid decreases metabolism in general and specially in the heart, delaying cardiac muscle repair and replacement. That leads to an enlarged flabby heart and often a viscous pericardial effusion. In hyperthyroid conditions, the cardiac muscles are overworked due to sinus tachycardia, increased demand for more cardiac output and associated hypertension. As the heart fails, the cardiac chambers dilate.
II. Hypertrophic Cardiomyopathy.
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant inherited disease. One copy of the mutated gene is all that is needed for HCM to manifest. Of the several genes responsible for HCM. These gene mutations are common- MYH7 and MYPPC3 gene; other mutated genes are MYPBC3, TNNT2, TNN13, TPN1, MLC2, and MLC3. 1 in 700 people in the Americas are carriers of these genes and about 75,000 people have HCM at a given time. But most are unaware of the presence of mutant genes, and unfortunately, a sudden cardiac arrest may be the first sign of it.
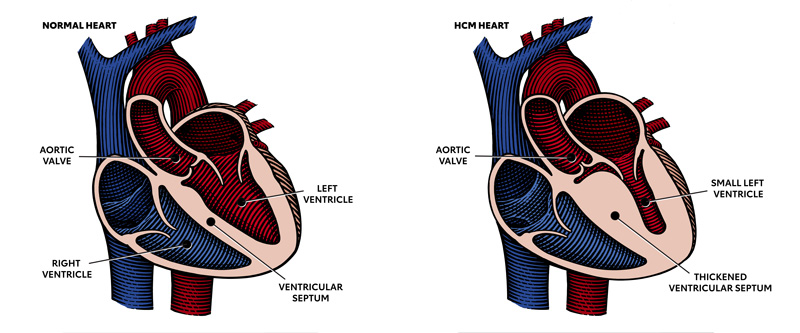
In the right-hand picture please note the thickened partition between the ventricular cavities. That part blocks the path of blood going out into the aorta during the ventricular ejection phase. In addition, the Mital valve is displaced and often deformed producing mitral insufficiency and reducing left ventricular ejection fraction further.
III. Restrictive Cardiomyopathy.
Restrictive cardiomyopathy is much less prevalent and accounts for only 5 % of all cases of diagnosed cardiomyopathies. This entity also consists of varied clinical conditions. The common pathophysiology is that abnormal proteins or abnormal cells accumulate between the muscle fibers of the ventricular wall, making the ventricular muscles less pliable and muscles fail to stretch fully. Returning blood accumulates in the atrium and causing both atria to dilate. The cardiac output falls and biventricular failure develops. In addition, the stagnant blood in heart chambers may clot and produce systemic and pulmonary embolism, which are additional features of restrictive cardiomyopathy.
Restrictive cardiomyopathy is a disease of the older generation. Shortness of breath and cardiac arrhythmia are presenting symptoms, additional symptoms are marked loss of weight, fatigue and the effects of arterial embolism like strokes, renal infarction, or limb ischemia.
1. In the Western world, the common causes of restrictive cardiomyopathies are Amyloidosis, Systemic sclerosis, Sarcoidosis and Hemochromatosis. Incidence is increasing in cases of post radiation therapy for malignancy and cancer treatment with Adriamycin.
2, Primary Endomyocardial Fibrosis and Restrictive Cardiomyopathy.
Taking the world as a whole, Primary Endomyocardial Fibrosis (EMF) is the most prevalent in this group and affects 12 million people. Most cases are seen in equatorial Africa and less frequently in tropical and subtropical Asia and in South America. EMF is somewhat similar to Loeffler eosinophilic endocarditis fibrosis seen in Non-tropical countries. The role of the eosinophil in EMF is still debated, some believe that eosinophils infiltrate the heart muscles due to dead and dying cardiac muscles. Others believe cytokines released by eosinophils produce myocardial necrosis and fibrosis.
The primary EMF is inherited as an autosomal dominant trait with variable penetration. Mutation of genes encoding Sarcomeric proteins - Troponin I, Troponin T alpha & beta, cardiac actin-myosin heavy chain are responsible for this disease.
[ For more information see footnote ]
IV. Arrhythmogenic Cardiomyopathy.
Arrhythmogenic cardiomyopathy is suspected to be present in 1 in 5,000 people, many of whom are asymptomatic. It is an inherited autosomal dominant condition. Mutation of at least 13 genes is identified. These genes are called Desmosomal genes and also called PKP2 genes. They provide instructions for making components of cell structures called Desmosomes. Under normal conditions, desmosomes keep the muscle fibers of the heart bound together so that all the heart muscles receive cardiac impulses from the sinus node in an orderly fashion and the ventricles can contract in unison.
In this disorder, the cells of the myocardium detach from each other and die. The damaged heart muscles are replaced by fibrous tissue and disrupts cardiac impulse transmission, leads to arrhythmia. The right ventricle is the prime site of this pathology, but since 2008, the same pathology was identified in the left ventricular wall.
PKP2 gene mutated people develop symptoms between 20 and 30 years of age. Sudden syncopal attacks are a usual presentation. Cardiomegaly and heart failure follow the same patterns as other forms of cardiomyopathy. PKP2 gene mutation was detected in 60% of cases, the other genetic abnormality is under investigation.
____________________________________
Footnote:
For further reading, please look at blogs dated -
1. Amyloidosis …. dated August19, 2000.
2. Hemochromatosis.......Oct 18, 2020.
3. Radiation pneumonitis...... January 10, 2021.
4. Connective tissue & MCTD...... Dec 14, 2022.
5. Sarcoidosis. This is a multisystem disease of unknown cause. Lung lesions resemble pulmonary tuberculosis, and extensive lymph node engagement is a distinctive feature. Many vital organs like the heart, eyes, and liver are commonly affected. Diagnosis requires tissue biopsy demonstration of non-caseating granuloma like TB but no acid-fast organisms are present. Other important lab findings are elevated serum YKL-40, ACE and IL-2R.
6. Adriamycin. Adriamycin is a potent cardiotoxic drug. It permanently damages heart muscles and produces fibrosis.
*********************************************

No comments:
Post a Comment